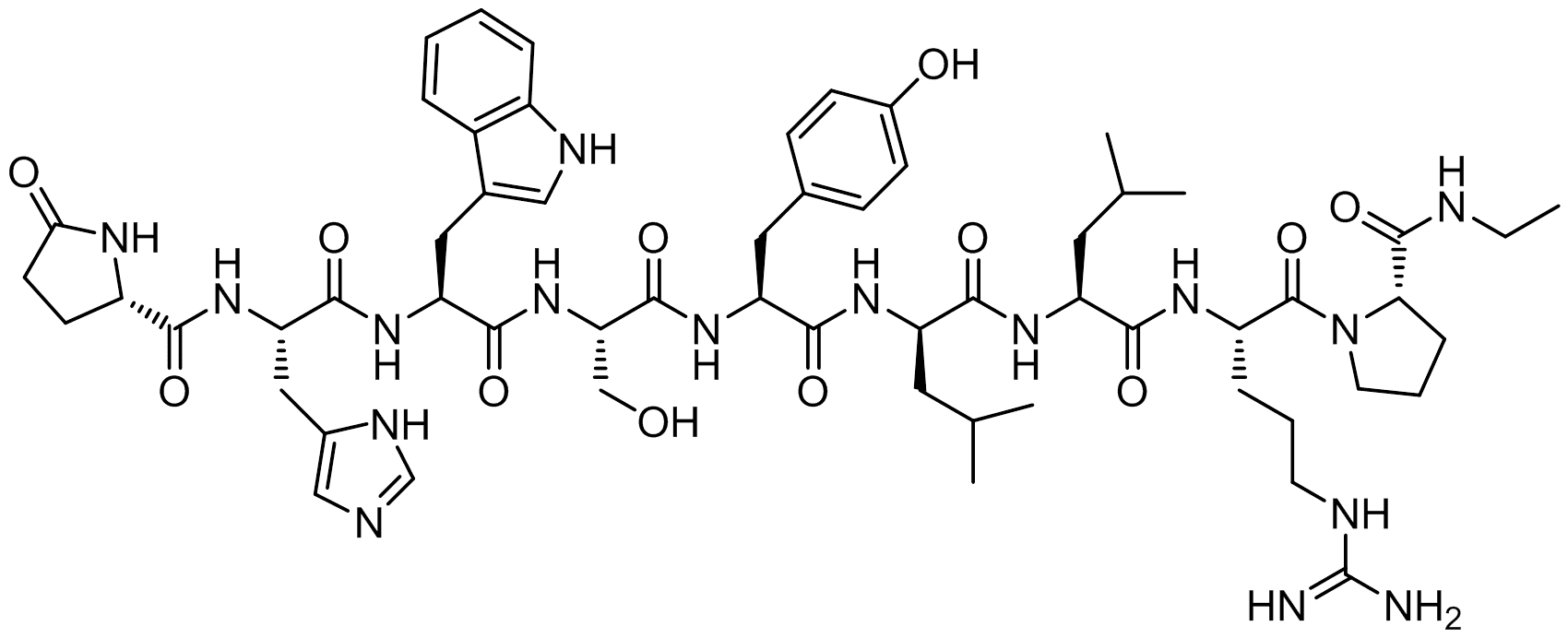
Leuprolide is a synthetic 9 residue peptide analog of gonadotropin releasing hormone belonging to the class of drugs called hormones or hormone antagonists. It is used to treat advanced prostate cancer, uterine fibroids, and endometriosis (under investigation for possible use in the treatment of mild to moderate Alzheimer’s disease). Leuprolide is also used to treat precocious puberty, control ovarian stimulation in in vitro fertilization (IVF), and for reducing sexual urges in pedophiles and other cases of paraphilia.
Indication
To treat prostate cancer, endometriosis, uterine fibroids, and premature puberty.
Pharmacodynamics
Leuprolide is a luteinizing hormone agonist that results in suppression of testicular or follicular steroidogenesis thus used in the palliative treatment of advanced prostate cancer.
Mechanism of Action
Leuprolide binds to the gonadotropin releasing hormone receptor and acts as an efficient inhibitor of gonadotropin secretion.
Metabolism
Primarily degraded by peptidase (instead of cytochrome P450 enzymes).
Clearance
Excretion in urine, 8.34 L/hour [healthy male receiving a 1-mg IV bolus]
Absorption
Bioavailability by subcutaneous administration is comparable to that by intravenous administration.
Chemistry and Manufacturing
The first industrial preparation for Leuprolide was introduced in 1977 by Fujino at Takeda Pharmaceutical Co. The patent describes two independent synthetic routes, a solid-phase (Boc/Bzl) and a solution phase route (Z/NO2). The solution route is better adapted for large scale manufacturing, described here, but requires isolation and characterization of the chemical intermediates throughout the process. The solution process involves few protection groups, only providing protection to the N-terminus (Z-protection, benzyloxycarbonyl) and guanidinium group of arginine (nitro, NO2) while leaving the side chains of histidine, tyrosine, serine, and tryptophan unprotected. Central to Fujino’s strategy are peptide fragments H-Leu-Arg(NO2)-Pro-NHCH2CH3 (1) and Pyr-His-Trp-Ser-Tyr-OH (2) which are condensed together to form leuprolide. Fragment 1 was prepared previously by Fujino/Takeda Pharm for the preparation of other LHRH analogs. Briefly, 1 is combined with Z-D-Leu-OH by way of N-hydroxy-5-norbornene-endo-2,3-dicarboxyimide (HONB) and N,N’-dicyclohexylcarbodiimide (DCC) activation to provide Z-D-Leu-Leu-Arg(NO2)-Pro-NHCH2CH3. Deprotection of the Z-group is accomplished with hydrobromic acid acetic acid solution (HBr-AcOH) to yield free amine H-D-Leu-Leu-Arg(NO2)-Pro-NHCH2CH3 (3). Fragment 2 is then activated with DCC-HONB and condensed with 3 to give full length nitro-protected leuprolide. Finally, the nitro-group is removed with aqueous formic acid and SnCl2.H2O to provide the final product, leuprolide.
Following Takeda Pharmaceutical’s 1977 process, a plethora of synthetic routes for leuprolide emerged that included solution-phase, solid-phase, and hybrid approaches. One highly convergent solution-phase methodology was introduced in 2011 by Kadzimirsz from Nanokem SA. Far from the minimalist approach of Takeda, this process is highly optimized with every functional group protected throughout the synthesis. While this process is more complex, it involves fewer side reactions and results in intermediates and final product with higher purity. The process begins with the synthesis of four dimer fragments, namely: (1) Boc-Arg(Mtr)-Pro-NHEt, (2) Boc-D-Leu-Leu-OAllyl, (3) Fmoc-Ser(tBu)-Tyr(tBu)-OAllyl, and (4) Pyr-His(Trt)-OMe. The first fragment is synthesized by the coupling of H-Pro-NHEt.HCl and Boc-Arg(Mtr)-OH (Mtr: 4-methoxy-2,3,6-trimethylbenzenesulfonyl) with HBTU and trimethylamine (TEA) in DMAC. The second fragment is made by combining pre-activated Boc-D-Leu-OSu and and H-Leu-OAll.TosOH (define OSu and tosylic acid). Fragment three, Fmoc-Ser(tBu)-Tyr(tBu)-OAllyl, is provided by the coupling of Fmoc-Ser(tBu)-OH and H-Tyr(tBu)-OAllyl with HBTU and diisopropylethylamine (DIPEA). Finally, H-His(Trt)-OMe.HCl, Pyr-OH, HBTU, and DIPEA are reacted to provide fragment four, Pyr-His(Trt)-OMe. The choices and variation of coupling reagents (HBTU vs. OSu), wide range of protection groups, different bases used in the activation steps (DIPEA vs TEA), and the unusual allyl ester protection of the C-terminus, demonstrates the high refinement level and cost optimization built into this process.
| Leuprolide sequence: pGlu-His-Trp-Ser-Tyr-D-Leu-Leu-Arg-Pro-NHEt |
||
| molecular weight: 1209.40 g/mol | molecular formula: C59H84N16O12 | US FDA Reg No.: 29355 |
| CAS No.: [53714-56-0] | Half-Life: approx. 3 hrs | CFDA Cert No.: ZJ20150025 |
|
||
Chemistry and Manufacturing Continued
The process continues with the deprotection of Boc-Arg(Mtr)-Pro-NHEt (1) with formic acid (HCO2H) to give free amine H-Arg(Mtr)-Pro-NHEt (1a). Separately, Boc-D-Leu-Leu-OAlly is saponified with aqueous sodium hydroxide (NaOH) in ethanol and then acidified to provide the corresponding free acid, Boc-D-Leu-Leu-OH (2a). 1a and 2a are then coupled together to provide tetramer Boc-D-Leu-Leu-Arg(Mtr)-Pro-NHEt (5).
The Fmoc group of Fmoc-Ser(tBu)-Tyr(tBu)-OAllyl (3) removed with base tris(2-aminoethyl)amine (TAEA) in ethylacetate (EtAc) to yield H-Ser(tBu)-Tyr(tBu)-OAllyl (3a). 3a is then coupled with Fmoc-Trp(Boc)-OH (HBTU/DIPEA) to provide trimer, Fmoc-Trp(Boc)-Ser(tBu)-Tyr(tBu)-OAllyl (6). Following Fmoc-deprotection (TAEA-EtAc), the free amine of 6 (6a) is combined (HBTU/DIPEA) with the saponified product of 4 (4b) to give tetramer Pyr-His(Trt)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-OAllyl (7). Saponification of 7 (NaOH(aq)-EtOH) followed by acidification provides free acid Pyr-His(Trt)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-OH (7a). The coupling of fragment 7a with amine 5a with PyBOP-HOBt gives fully protected leuprolide Pyr-His(Trt)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-Leu-Leu-Arg(Mtr)-Pro-NHEt (8). Final deprotection is accomplished with trifluoroacetic acid (TFA) and dithiothreitol (DTT) in dichloromethane and (CH2Cl2) to provide leuprolide.
While solution-phase routes by Takeda Pharmaceutical and Nanokem utilize very different protection schemes, both are designed for scalability (i.e., large-scale manufacturing). Most of the solid-phase production routes involve the use of 2-chlorotrityl chloride resin (2-CTC) or the equivalent (e.g., HMPB-AM) because of the need for mild cleavage conditions to maintain sidechain protection during C-terminal ethyl amidation. Some chemists may consider 2-CTC- and HMPB-AM-based preparations of leuprolide to all be hybrid approaches because the ethyl amidation step is carried out in solution after cleavage from the resin. For the purposes of this review, we will still consider these preparations to be solid-phase approaches. Solid-phase syntheses, in general, has the advantage of not requiring isolation of intermediates, excess reagents to drive reactions to completion, and the easy removal of excess reagents by iterative resin washes. While solid-phase synthesis provides these advantages, some economical disadvantages also exist. Excess reagents are a convenient way to ensure reaction completeness; however, excess reagents means additional costs of the building blocks. Excess solvents to wash the resin also adds cost to the process, both in the initial purchase and in waste disposal costs.
- Fujino, M., Fukuda, T. and Shinagawa, S., Takeda Pharmaceutical Co Ltd. “Nonapeptide amide analogs of luteinizing releasing hormone.” U.S. Patent 4,008,209, 1977, Feb 15.
- M Fujino, S Shinagawa, T Fukuda, S Kobayashi, M Obayashi. Takeda Pharmaceutical Co Ltd. “Novel nonapeptide amide analogs of luteinizing hormone releasing factor.” U.S. Patent 3,853,837, 1974, Dec 10.
- Zhou Daming. Shanghai Soho Yiming Pharmaceutical Co., Ltd. “Solid phase polypeptide synthesis preparation method for leuprorelin.” Patent CN1865280B, 2012, March 21.
- Srivastava, K.S. and Davis, M.R., Mallinckrodt LLC. Solid support for Fmoc-solid phase synthesis of peptides. U.S. Patent 7,714,063, 2010, May 11.
- Dae-young Kim, Jae Il Kim, Jin-kwon Kim, Young Soo Yoon. Anisen Co., Ltd. “Process for the Preparation of Leuprolide.” Patent KR101171095B1, 2012, Aug 3.
- Wang Caidian, Liu Biao, Gu Haitao, Xu Feng, Sun Meilu, Li Jiajun. Jiangsu Nuotai Biological Pharmaceutical Co., Ltd. “Leuprorelin synthesis method.” Patent Application No.:CN105330726A, 2016, Feb 17.
- Ananda Kuppanna, Bulli Raju Kamana, Sreelatha Vanjivaka, Debashish Datta. Mylan Laboratories Pvt Ltd . “Novel Process For The Preparation Of Leuprolide And Its Pharmaceutically Acceptable Salts Thereof.” US Patent Application US20130060004A1, 2013, March 3.
