FKC (FKCRRWQWRMKK-NH2), rhodamine-conjugated FKC (Rho-FKCRRWQWRMKK-NH2), and IDR (VQRWLIVWRIRK-NH2) peptides were synthesized using solid phase F-moc chemistry by CPC Scientific, Inc. with a purity greater than 95%.
Abstract
Tumor necrosis factor (TNF) receptor 1 (TNFR1) plays a pivotal role in mediating TNF induced downstream signaling and regulating inflammatory response. Recent studies have suggested that TNFR1 activation involves conformational rearrangements of preligand assembled receptor dimers and targeting receptor conformational dynamics is a viable strategy to modulate TNFR1 signaling. Here, we used a combination of biophysical, biochemical, and cellular assays, as well as molecular dynamics simulation to show that an anti-inflammatory peptide (FKCRRWQWRMKK), which we termed FKC, inhibits TNFR1 activation allosterically by altering the conformational states of the receptor dimer without blocking receptor–ligand interaction or disrupting receptor dimerization. We also demonstrated the efficacy of FKC by showing that the peptide inhibits TNFR1 signaling in HEK293 cells and attenuates inflammation in mice with intraperitoneal TNF injection. Mechanistically, we found that FKC binds to TNFR1 cysteine-rich domains (CRD2/3) and perturbs the conformational dynamics required for receptor activation. Importantly, FKC increases the frequency in the opening of both CRD2/3 and CRD4 in the receptor dimer, as well as induces a conformational opening in the cytosolic regions of the receptor. This results in an inhibitory conformational state that impedes the recruitment of downstream signaling molecules. Together, these data provide evidence on the feasibility of targeting TNFR1 conformationally active region and open new avenues for receptor-specific inhibition of TNFR1 signaling.
SOCIAL MEDIA
Connect with us and stay updated by following our social media channels.
Latest Briefings from our Knowledge Center
Press Releases, Industry News, Articles, and Technical Content

Gain insights from Joseph Denby's rapid-fire Q&A spotlight at NextGen Biomed, where he explores the landscape of peptide and oligo APIs, as well as the role of green chemistry in manufacturing.

Home / Knowledge Center / Articles
Highlights from CPC Scientific’s 2025 Annual Conference
GMP Peptide Manufacturing
We’re excited to share the key moments from CPC Scientific’s 2025 Annual Conference!
Hosted in San Jose, California, our annual gathering brought together colleagues from various teams and timezones for meaningful updates, focused […]

At NextGen Biomed 2025, Joseph Denby delivered a cutting-edge presentation highlighting the critical role green chemistry plays in the sustainable production of peptide APIs and peptide–oligo conjugates...

Home / Knowledge Center / Articles
CPC Scientific at DCAT 2025!
GMP Peptide Manufacturing
What an amazing time we had at DCAT 2025! It was fantastic to meet with colleagues, clients, and friends, and showcase CPC Scientific’s cutting-edge capabilities in synthetic API development and commercial production.
A heartfelt […]

We are excited to share that CPC Scientific Inc. has officially received the Good Manufacturing Practice (GMP) Certificate in accordance with the ISO 22716:2007(E) Cosmetic Guidelines.
This certification highlights our unwavering commitment to delivering the highest quality peptide ingredients for cosmetic use, guaranteeing:
- Superior Quality Control – Striving for excellence in safety, consistency, and purity.
- Global […]

In Part 1 of our Minimal Protection Group Strategies for SPPS, we discussed methods for eliminating sidechain protection on hydroxy-bearing amino acids such as serine, threonine, tyrosine, and hydroxyproline. By omitting t-butyl protection, we enhanced atom economy and avoided the use of hazardous solvents typically required to remove these protection groups. In Part 2, we present a new case study, expanding our approach to include the unprotected side chains of histidine, tryptophan, and arginine. We demonstrate the synthesis of a Goserelin peptide API impurity, showcasing how a convergent peptide fragment strategy can be used to eliminate the need for TFA and diethyl ether, eliminate side chain protection of Arginine, Histidine, and Tryptophan.
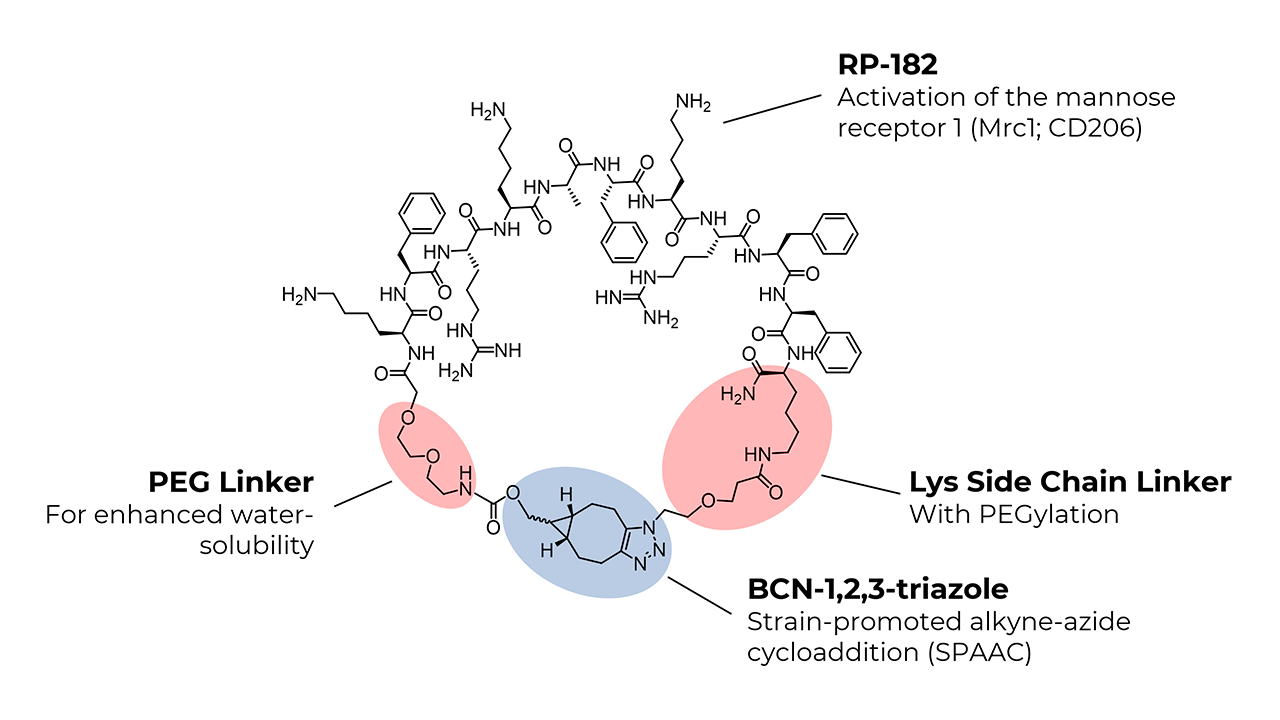
The synthesis of the linear RP-182 analog, bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl-PEG2-Lys-Phe-Arg-Lys-Ala-Phe-Lys-Arg-Phe-Phe-Lys(azido-PEG)-NH2, was achieved using standard solid-phase peptide synthesis (SPPS) protocols. After cleaving the linear peptide from the resin, macrocyclization was performed in the liquid phase through a strain-promoted click reaction. BCN introduces extra ring strain due to its fused cyclopropane structure. The combined effect of ring strain, the selection of BCN, and copper catalysis significantly increases the macrocyclization efficiency of longer peptides like RP-182.
Nakagawa, Mayumi, Teresa Evans, Milan Bimali, Hannah Coleman, Jasmine Crane, Nadia Darwish, Jennifer L. Faulkner et al. MedRxiv (2025): 2025-01.
The vaccine consisted of four current good manufacturing production-grade synthetic peptides covering the HPV 16 E6 protein [amino acid (aa)1-45, 46-80, 81-115, and 116-158] (CPC Scientific, San Jose, CA [..]
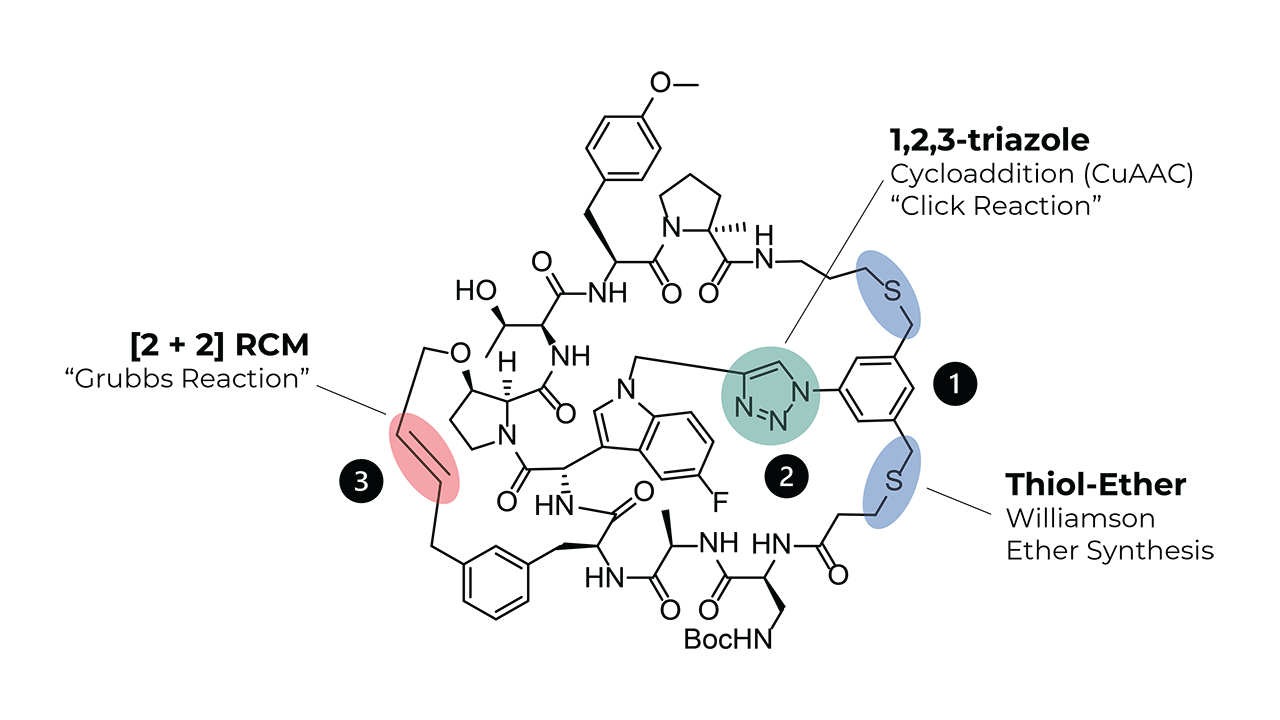
Stapled peptides have emerged as a powerful tool in drug discovery and therapeutic development due to their ability to overcome the limitations associated with traditional peptide drugs, such as poor stability and low cell permeability. By introducing staples into the peptide backbone, researchers can stabilize peptide conformations and enhance their interactions with target proteins, resulting in improved efficacy and specificity. This approach not only addresses the challenges of peptide drug design but also opens new avenues for targeting challenging biomolecular interactions that are difficult to modulate with small molecules or antibodies. The development of stapled peptides has led to significant advancements in targeting protein-protein interactions, addressing previously intractable diseases, and enhancing the precision of therapeutic interventions.
